
Key learning objectives
Understand the role of insulin as a metabolic hormone
Find out how insulin regulates glucose metabolism
Discuss the specific action of insulin in different tissues
Understand the importance of insulin sensitivity for a healthy aging process
What Is Insulin?
Insulin is a hormone produced by beta cells in the pancreas with a central role in the regulation of metabolism and cell energy reserves. The major metabolic action of insulin is to regulate blood glucose levels and to promote the storage of energy substrates as macromolecules that can be mobilized between meals or in contexts of high energy demand.
Insulin acts on different metabolic tissues to promote the replenishment of energy stores: it stimulates glucose uptake from the blood by the liver and skeletal muscle and its storage as glycogen; it stimulates the uptake of fatty acids by adipose tissue and their storage as triglycerides; and it stimulates amino acid uptake by muscles and their use for muscle protein synthesis. Therefore, insulin is a major anabolic hormone [1].
[Anabolism is the set of metabolic pathways that build large molecules from smaller molecules and store cell energy in their chemical bonds. The opposite processes, i.e., the degradative pathways that break down large molecules into smaller molecules, releasing the energy stored in the chemical bonds, are known as catabolism.]
Insulin is an important metabolic hormone that stimulates the storage of energy reserves in tissues in the form of large molecules.
Insulin Regulates Blood Glucose Levels
One of the major actions of insulin, and that for which insulin is most known, is the regulation of blood glucose levels [1]. Following a meal, insulin secretion by the pancreas is stimulated by the rise in blood glucose levels, which thus acts as a signal for insulin secretion. In turn, the rise in blood insulin levels acts as a signal for glucose uptake from the blood and into tissues. When blood glucose levels return to homeostatic values, insulin secretion by the pancreas diminishes.

Figure 1: Insulin and blood glucose levels. Source: OpenStax,
Biology for AP® Courses; 24.3 Homeostasis. License: CC BY 4.0.
Insulin stimulates glucose uptake by mobilizing glucose transporters (GLUTs) to the cell membrane, which are required for glucose to cross the cell membrane. There are different types of GLUTs with different properties. GLUT4 is an insulin-dependent glucose transporter, meaning that it takes up glucose when it’s activated by insulin [1]. GLUT4 is mainly expressed in adipose tissue, skeletal muscle cells, and cardiac muscle cells. In these tissues, which are the major energy storage tissues, the uptake of glucose from the blood to the cells is therefore stimulated by insulin. Insulin-induced GLUT4 activity in metabolic tissues is important because it determines the rate of glucose uptake from the blood into tissues and therefore plays a key role in glucose homeostasis and in the rate of glucose metabolism [2].
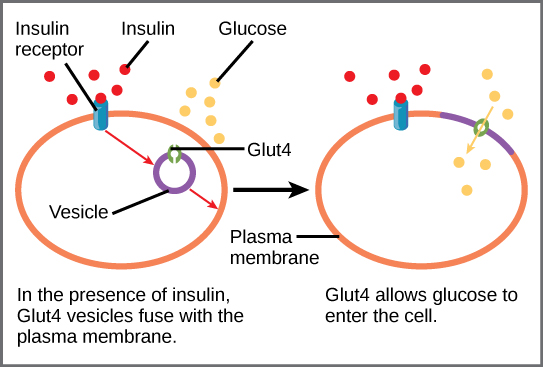
Figure 2: Insulin-dependent glucose uptake via GLUT4.
Source: OpenStax, Biology for AP® Courses;
7.7 Regulation of Cellular Respiration. License: CC BY 4.0
The other types of glucose transporters, GLUT1, GLUT2, and GLUT3, are independent of insulin signaling. GLUT1 and GLUT3 are expressed in all tissues and are responsible for basal glucose uptake. GLUT2 is expressed in pancreatic beta-cells and liver cells and has properties that allow glucose to enter these tissues at a higher rate when blood glucose levels rise.
Insulin stimulates glucose uptake by skeletal muscle cells, adipose tissue, and cardiac muscle cells by recruiting the GLUT4 glucose transporter in these metabolic tissues.
Qualia Life (previously called Eternus) contains a set of ingredients in its formulation that support the expression of the glucose transporter GLUT4. These include Sophora japonica L. Flower Extract (95% Rutin) [3], Citrus sinensis L. Fruit Extract (40% Nobiletin) [4,5], Cinnamon extract (Cinnulin PF®) [6–8], Grape Proanthocyanidins (from BioVin® French Red Grapes Extract) [9,10], Lipoic Acid [11–13], and Inositol (as Myo-Inositol + D-Chiro-Inositol) [14–16].*
Insulin Promotes Glucose Storage In The Liver
The liver doesn’t need insulin to take up glucose. When blood glucose levels are high, glucose enters the liver at a higher rate through the GLUT2 transporter [17]. The general action of insulin in the liver is to promote glucose storage as glycogen and to suppress hepatic glucose production when blood glucose levels are high. Insulin promotes the synthesis of glycogen (i.e., glycogenesis) by stimulating the activity of the enzyme glycogen synthase. At the same time, insulin inhibits glucose release from glycogen breakdown (i.e., glycogenolysis) and glucose production from non-carbohydrate precursors (i.e., gluconeogenesis) [18].
The liver is the major glycogen storage organ per weight, with glycogen making up around 10% of the liver’s weight. In the liver, glycogen production and degradation are regulated to maintain blood-glucose levels as required to meet the needs of our body as a whole. The net effect of insulin in the liver is the replenishment of glycogen stores that can be mobilized when blood glucose levels decrease—between meals, for example—to maintain stable blood glucose levels [1].
Our body stores about a day’s worth of glycogen; when these stores are replenished, excess glucose is converted into fatty acids and then into triglycerides. Insulin also decreases fatty acid oxidation and increases fatty acid and triglyceride synthesis as part of an energy-storage and biosynthesis-promoting effect [19].
Insulin stimulates the creation of liver glucose stores in the form of glycogen and suppresses the production and release of glucose from the liver to the blood when glucose levels are high.
Insulin Promotes Glucose and Protein Storage In Muscles
Skeletal muscle is the other major site of glycogen storage, along with the liver. Although the liver stores more glycogen per weight (10% in the liver versus 2% in skeletal muscle), more glycogen is stored in skeletal muscles overall because of their greater mass—about 40% of total body mass. Therefore, muscle is the largest tissue target of insulin, accounting for around 70% of whole-body insulin-stimulated glucose uptake [19].
In skeletal muscle, insulin stimulates the uptake of glucose via GLUT4, promotes glycogen synthesis, and inhibits glycogen breakdown. When muscle glycogen storage is replete, glucose utilization is maintained by converting it to lactate, which can be released into the blood and taken up by the liver to be converted to glycogen (an “indirect pathway” of glycogen synthesis). This allows for the removal of excess blood glucose by muscles even when muscle glycogen stores are full, thereby helping to maintain normal blood glucose levels.
In muscles, glycogen synthesis and degradation are regulated to meet the energy needs of the muscle [20]. Insulin promotes the storage of glycogen to be used as a source of glucose for energy production. Because ATP production from glucose is faster, glucose is the preferred fuel during high-intensity bursts of exercise. But during mild to moderate intensity prolonged exercise, fatty acids become the preferred fuel because, even though ATP production by fatty acid oxidation is slower, it yields more ATP. Insulin also promotes the uptake of free fatty acids from the blood by muscles to be used as fuel or stored as triglycerides [19]. But muscles only store a small amount of triglycerides; the vast majority of fatty acids used by muscles come from triglyceride breakdown in adipose tissue.
Insulin promotes the uptake of glucose and its storage as glycogen in skeletal muscles. Muscles are the body’s major glycogen reserve.
Insulin also has important effects on protein metabolism in muscles, where it promotes the uptake of amino acids, stimulates protein synthesis, and decreases protein degradation. By doing so, insulin stimulates muscle growth and modulates skeletal muscle fiber regeneration and growth. Insulin also activates and promotes the proliferation and differentiation of muscle satellite cells (muscle precursor cells) [21,22]. Therefore, a major effect of insulin in protein metabolism is to favor the buildup of muscle protein [1].
Insulin Promotes Fatty Acid Storage In Adipose Tissue
Adipose tissue accounts for a relatively small proportion (<10%) of peripheral glucose utilization in response to insulin. Nevertheless, it is very sensitive to insulin, which stimulates glucose uptake via GLUT4. In adipose cells, although some glucose is stored as glycogen, most is converted into lipids [19].
In adipose tissue, insulin’s major effects are on lipid metabolism. Insulin stimulates the uptake of fatty acids from the blood, stimulates fatty acid and triglyceride synthesis (i.e., lipogenesis), and decreases the rate of triglyceride breakdown (i.e., lipolysis). Insulin thereby lowers the blood levels of fatty acids and promotes energy storage in adipose tissue. Through the effect of insulin, adipose tissue regulates the levels of fatty acids in the blood following a meal [23].
Adipose tissue is the major store of triglycerides in the body. When necessary for energy production, triglycerides are mobilized and broken down to fatty acids, which are carried via the blood to other tissues, particularly muscles, where they can be metabolized through fatty acid oxidation to produce energy [19].
In adipose tissue, insulin stimulates the uptake of fatty acids from the blood and its storage as triglycerides.

Figure 3: Insulin action in metabolic tissues.
Source: OpenStax, Anatomy and Physiology;
24.5 Metabolic States of the Body. License: CC BY 4.0
Insulin Action In The Brain Promotes Energy Homeostasis
Insulin’s effects on energy metabolism are not limited to its direct action in metabolic tissues, they are also a result of insulin signaling in the central nervous system. The brain is also a target for insulin, where it modulates brain glucose sensing and the brain’s responses to blood glucose levels [24].
Brain insulin influences glucose homeostasis and whole-body energy balance by regulating appetite, for example. Insulin decreases food intake by decreasing the expression of orexigenic (i.e., appetite stimulant) neuropeptides and increasing the expression of anorexigenic (i.e., appetite inhibitor) neuropeptides [25]. Therefore, insulin has an appetite-inhibiting effect.
Through its action in the brain, insulin also influences energy expenditure through the regulation of body temperature. Insulin acting in the brain can activate heat-producing mechanisms in brown adipose tissue and enhance diet-induced thermogenesis [24,26,27].
Furthermore, brain insulin signaling modulates a number of neurotransmitter systems and is essential for neurological processes, participating in the regulation of cognition, learning, memory, and mood [28].
Brain insulin signaling influences energy homeostasis and whole-body energy balance by regulating appetite and energy expenditure.
What Is Insulin Sensitivity?
Insulin sensitivity is the measure of a tissue’s ability to efficiently respond to insulin and its actions. Loss of insulin sensitivity decreases the ability of insulin-sensitive tissues to take up glucose. As a result, blood glucose levels remain excessively high, a state known as hyperglycemia. Also, to make up for the loss of insulin sensitivity, insulin production is prolonged, which increases blood levels of insulin, a state known as hyperinsulinemia. Loss of insulin sensitivity is known as insulin resistance [18].
Insulin sensitivity tends to diminish with age, but lifestyle patterns play a very important part in the development of insulin resistance. Overnutrition, excessive weight, and insufficient physical activity are among the major contributors to the development of insulin resistance[18]. In experiments that restrict sleep (i.e., produce sleep deprivation), insulin sensitivity is decreased [29–32], while extending sleep in adults who consistently get insufficient sleep improves insulin sensitivity [33].
Insulin sensitivity—the measure of a tissue’s ability to efficiently respond to the actions of insulin—is a hallmark of healthy longevity.
One of the major risk factors for insulin resistance is ectopic fat accumulation, i.e., fat accumulation in abnormal places, particularly in muscles and in the liver [18,34,35]. Ectopic fat accumulation is associated with increased levels of lipid intermediate metabolites that disrupt insulin signaling [18,36]. Ectopic fat accumulation can be driven by age-related decreases in mitochondrial function and fatty acid oxidation capacity due to an accumulation of oxidative damage [36]. But it can also be caused or aggravated by overnutrition and a sedentary lifestyle.
Muscles provide a good example of how overeating and physical inactivity affect insulin sensitivity: muscle fatty acid uptake increases with higher levels of fatty acids in the blood, which in turn increase with overeating and adiposity[37]; and muscle fatty acid oxidation capacity is dependent on physical activity and increases with endurance training[38]. This means that overeating and physical inactivity increase muscle fatty acid uptake, but diminish fatty acid oxidation capacity, which results in ectopic fat accumulation and insulin resistance in muscles. By opposition, decreased fatty acid levels and improved muscle fatty acid oxidation capacity have been shown to improve insulin sensitivity [39,40].
Because skeletal muscle has a key role in regulating blood glucose levels in response to insulin, muscle insulin resistance has a great impact on blood glucose levels.
How Insulin Resistance Affects Healthy Aging
Insulin resistance can easily escalate to serious tissue dysfunctions. For example, if glucose uptake by muscles decreases due to insulin resistance, glucose may be shunted to the liver; there, if glycogen stores are replete, glucose will be converted into fatty acids and triglycerides, which will then be exported for storage in adipose tissue. An exaggerated production of triglycerides in the liver can lead to local ectopic fat accumulation, known as fatty liver or hepatic steatosis. The associated accumulation of lipid intermediate metabolites can disrupt insulin signaling, feeding the progression of insulin resistance and further accumulation of ectopic fat [18,41].
As these metabolic changes start to unfold, fat accumulation in adipose tissue also increases. This results in an increased secretion of inflammatory mediators by adipose tissue, which also promotes insulin resistance [34,42–45].
Insulin-resistant adipose tissue loses the ability to effectively regulate lipid levels in the blood. As a consequence, all tissues become chronically exposed to higher concentrations of fatty acids and triglycerides in the blood. An increase in blood lipid levels increases the uptake of fatty acids by muscles and the liver, leading to further lipid accumulation and enhanced insulin resistance [18,23].
Insulin-resistant adipose tissue loses ability to effectively regulate lipid levels in the blood.
Therefore, loss of insulin sensitivity in skeletal muscles, the liver, or adipose tissue, can easily trigger a feed-forward loop of events that will increase, perpetuate, and generalize insulin resistance. The associated release of inflammatory mediators into the blood can cause a state of chronic low-grade inflammation that affects all tissues in the body and that also feeds the loop of insulin resistance, fat accumulation, and inflammation. These processes also trigger a cascade of events that increase the production of reactive oxygen species (ROS), which in turn lead to DNA damage, mitochondrial dysfunction, and impaired cellular functions [34,42–45].
Insulin resistance is associated with processes that negatively impact the aging process and contribute to many age-related dysfunctions.
Why Supporting Insulin Signaling Is Important
Insulin influences all aspects of human health by supporting many molecular and cellular functions. Maintenance of insulin sensitivity is a hallmark of healthy longevity. In fact, maintenance of insulin sensitivity may be one of the secrets to “successful aging” as high insulin sensitivity and low levels of insulin have been associated with increased longevity in several studies [46,47]. Importantly, studies with healthy centenarians have shown that high insulin sensitivity is a characteristic of these individuals, being a probable contributor to their extreme lifespan [48,49].
By opposition, insulin resistance is associated with a decrease in longevity for several reasons: it increases the development of cellular impairments that accelerate the aging process [50]; it is a risk factor for poor cardiometabolic health (i.e., age-related issues that impact heart health and metabolic performance) [51]; and it has been linked to unhealthy brain aging [52].
Supporting healthy insulin signaling was a major emphasis in the formulation of Qualia Life.* It includes a range of ingredients that may support the maintenance of healthy insulin sensitivity. These include Strawberry Seed Extract [53], Cocoa Seed Extract [54–60], Rosmarinus officinalis Leaf Extract (50% ursolic acid) [61–63], Sensoril® Ashwagandha Withania somnifera Root and Leaf Extract [64–66], Apigenin (from Citrus grandis Fruit Extract) [67–69], Gynostemma pentaphyllum [70–72], Vitamin K2 as Menaquinone-7 [73–75], Sophora japonica L. Flower Extract (95% Rutin) [3,76,77], Citrus sinensis L. Fruit Extract (40% Nobiletin) [5,78,79], Cinnamon extract (Cinnulin PF®) [80–82], β-Hydroxy-β-Methylbutyric acid (HMB) [83], Grape Proanthocyanidins (from BioVin® French Red Grapes Extract) [9,84,85], Resveratrol (from BioVin® French Red Grapes Extract) [86–88], L-Carnitine [89–91], Lipoic Acid [12,92,93], and Inositol (as Myo-Inositol + D-Chiro-Inositol) [94–96].*
*These statements have not been evaluated by the Food and Drug Administration. This product is not intended to diagnose, treat, cure, or prevent any disease. These statements are not intended as general medical advice. This product is not a replacement for prescription medication. Please consult your physician before taking any dietary supplements.
References
[1] J.M. Berg, J.L. Tymoczko, G.J. Gatto, L. Stryer, eds., Biochemistry, 8th ed, W.H. Freeman and Company, 2015.
[2] J.E. Hall, Guyton and Hall Textbook of Medical Physiology, Elsevier Health Sciences, 2015.
[3] C.-Y. Hsu, H.-Y. Shih, Y.-C. Chia, C.-H. Lee, H. Ashida, Y.-K. Lai, C.-F. Weng, Mol. Nutr. Food Res. 58 (2014) 1168–1176.
[4] Y.-S. Lee, B.-Y. Cha, S.-S. Choi, B.-K. Choi, T. Yonezawa, T. Teruya, K. Nagai, J.-T. Woo, J. Nutr. Biochem. 24 (2013) 156–162.
[5] Y.-S. Lee, B.-Y. Cha, K. Saito, H. Yamakawa, S.-S. Choi, K. Yamaguchi, T. Yonezawa, T. Teruya, K. Nagai, J.-T. Woo, Biochem. Pharmacol. 79 (2010) 1674–1683.
[6] Y. Shen, N. Honma, K. Kobayashi, L.N. Jia, T. Hosono, K. Shindo, T. Ariga, T. Seki, PLoS One 9 (2014) e87894.
[7] X. Sheng, Y. Zhang, Z. Gong, C. Huang, Y.Q. Zang, PPAR Res. 2008 (2008) 581348.
[8] Y. Shen, M. Fukushima, Y. Ito, E. Muraki, T. Hosono, T. Seki, T. Ariga, Biosci. Biotechnol. Biochem. 74 (2010) 2418–2425.
[9] G.F. da Costa, I.B. Santos, G.F. de Bem, V.S.C. Cordeiro, C.A. da Costa, L.C.R.M. de Carvalho, D.T. Ognibene, A.C. Resende, R.S. de Moura, Phytother. Res. 31 (2017) 1621–1632.
[10] M. Aoun, F. Michel, G. Fouret, A. Schlernitzauer, V. Ollendorff, C. Wrutniak-Cabello, J.-P. Cristol, M.-A. Carbonneau, C. Coudray, C. Feillet-Coudray, Br. J. Nutr. 106 (2011) 491–501.
[11] Y. Wang, X. Li, Y. Guo, L. Chan, X. Guan, Metabolism 59 (2010) 967–976.
[12] A. Rudich, A. Tirosh, R. Potashnik, M. Khamaisi, N. Bashan, Diabetologia 42 (1999) 949–957.
[13] M. Khamaisi, R. Potashnik, A. Tirosh, E. Demshchak, A. Rudich, H. Tritschler, K. Wessel, N. Bashan, Metabolism 46 (1997) 763–768.
[14] A. Yap, S. Nishiumi, K.-I. Yoshida, H. Ashida, Cytotechnology 55 (2007) 103–108.
[15] N.T. Dang, R. Mukai, K.-I. Yoshida, H. Ashida, Biosci. Biotechnol. Biochem. 74 (2010) 1062–1067.
[16] Y. Yamashita, M. Yamaoka, T. Hasunuma, H. Ashida, K.-I. Yoshida, J. Agric. Food Chem. 61 (2013) 4850–4854.
[17] M. Tal, Y. Liang, H. Najafi, H.F. Lodish, F.M. Matschinsky, J. Biol. Chem. 267 (1992) 17241–17247.
[18] V.T. Samuel, G.I. Shulman, J. Clin. Invest. 126 (2016) 12–22.
[19] J. Zhang, F. Liu, IUBMB Life 66 (2014) 485–495.
[20] G. Dimitriadis, P. Mitrou, V. Lambadiari, E. Maratou, S.A. Raptis, Diabetes Res. Clin. Pract. 93 (2011) S52–S59.
[21] A. Philippou, M. Maridaki, A. Halapas, M. Koutsilieris, In Vivo 21 (2007) 45–54.
[22] R.P. Rhoads, L.H. Baumgard, S.W. El-Kadi, L.D. Zhao, J. Anim. Sci. 94 (2016) 1791–1802.
[23] K.N. Frayn, Diabetologia 45 (2002) 1201–1210.
[24] A. Kleinridders, H.A. Ferris, W. Cai, C.R. Kahn, Diabetes 63 (2014) 2232–2243.
[25] M.W. Schwartz, S.C. Woods, D. Porte Jr, R.J. Seeley, D.G. Baskin, Nature 404 (2000) 661–671.
[26] M.D. Sharma, A.J. Garber, J.A. Farmer, Endocr. Pract. 14 (2008) 373–380.
[27] C. Benedict, S. Brede, H.B. Schiöth, H. Lehnert, B. Schultes, J. Born, M. Hallschmid, Diabetes 60 (2011) 114–118.
[28] S.-H. Lee, J.M. Zabolotny, H. Huang, H. Lee, Y.-B. Kim, Mol Metab 5 (2016) 589–601.
[29] M. González-Ortiz, E. Martínez-Abundis, B.R. Balcázar-Muñoz, S. Pascoe-González, Diabetes Nutr. Metab. 13 (2000) 80–83.
[30] O.M. Buxton, M. Pavlova, E.W. Reid, W. Wang, D.C. Simonson, G.K. Adler, Diabetes 59 (2010) 2126–2133.
[31] M.D. Robertson, D. Russell-Jones, A.M. Umpleby, D.-J. Dijk, Metabolism 62 (2013) 204–211.
[32] J.L. Broussard, D.A. Ehrmann, E. Van Cauter, E. Tasali, M.J. Brady, Ann. Intern. Med. 157 (2012) 549–557.
[33] R. Leproult, G. Deliens, M. Gilson, P. Peigneux, Sleep 38 (2015) 707–715.
[34] V.T. Samuel, G.I. Shulman, Cell 148 (2012) 852–871.
[35] L.P. Turcotte, J.S. Fisher, Phys. Ther. 88 (2008) 1279–1296.
[36] K.F. Petersen, D. Befroy, S. Dufour, J. Dziura, C. Ariyan, D.L. Rothman, L. DiPietro, G.W. Cline, G.I. Shulman, Science 300 (2003) 1140–1142.
[37] G. Boden, Endocrinol. Metab. Clin. North Am. 37 (2008) 635–46, viii–ix.
[38] J. Achten, A.E. Jeukendrup, Nutrition 20 (2004) 716–727.
[39] F. Shojaee-Moradie, K.C.R. Baynes, C. Pentecost, J.D. Bell, E.L. Thomas, N.C. Jackson, M. Stolinski, M. Whyte, D. Lovell, S.B. Bowes, J. Gibney, R.H. Jones, A.M. Umpleby, Diabetologia 50 (2007) 404–413.
[40] G. Perdomo, S.R. Commerford, A.-M.T. Richard, S.H. Adams, B.E. Corkey, R.M. O’Doherty, N.F. Brown, J. Biol. Chem. 279 (2004) 27177–27186.
[41] F. Magkos, X. Su, D. Bradley, E. Fabbrini, C. Conte, J.C. Eagon, J.E. Varela, E.M. Brunt, B.W. Patterson, S. Klein, Gastroenterology 142 (2012) 1444–6.e2.
[42] C. de Luca, J.M. Olefsky, FEBS Lett. 582 (2008) 97–105.
[43] L. Chen, R. Chen, H. Wang, F. Liang, Int. J. Endocrinol. 2015 (2015) 508409.
[44] H. Yaribeygi, F.R. Farrokhi, A.E. Butler, A. Sahebkar, J. Cell. Physiol. (2018).
[45] M.H. Park, D.H. Kim, E.K. Lee, N.D. Kim, D.S. Im, J. Lee, B.P. Yu, H.Y. Chung, Arch. Pharm. Res. 37 (2014) 1507–1514.
[46] C.A. Wijsman, M.P. Rozing, T.C.M. Streefland, S. le Cessie, S.P. Mooijaart, P.E. Slagboom, R.G.J. Westendorp, H. Pijl, D. van Heemst, Leiden Longevity Study group, Aging Cell 10 (2011) 114–121.
[47] N.M. Templeman, S. Flibotte, J.H.L. Chik, S. Sinha, G.E. Lim, L.J. Foster, C. Nislow, J.D. Johnson, Cell Rep. 20 (2017) 451–463.
[48] G. Paolisso, A. Gambardella, S. Ammendola, A. D’Amore, V. Balbi, M. Varricchio, F. D’Onofrio, Am. J. Physiol. 270 (1996) E890–4.
[49] M. Barbieri, A. Gambardella, G. Paolisso, M. Varricchio, Exp. Gerontol. 43 (2008) 74–78.
[50] Z. Cheng, Y. Tseng, M.F. White, Trends Endocrinol. Metab. 21 (2010) 589–598.
[51] H. Gill, M. Mugo, A. Whaley-Connell, C. Stump, J.R. Sowers, Am. J. Med. Sci. 330 (2005) 290–294.
[52] M. Schubert, D. Gautam, D. Surjo, K. Ueki, S. Baudler, D. Schubert, T. Kondo, J. Alber, N. Galldiks, E. Küstermann, S. Arndt, A.H. Jacobs, W. Krone, C.R. Kahn, J.C. Brüning, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 3100–3105.
[53] T. Goto, A. Teraminami, J.-Y. Lee, K. Ohyama, K. Funakoshi, Y.-I. Kim, S. Hirai, T. Uemura, R. Yu, N. Takahashi, T. Kawada, J. Nutr. Biochem. 23 (2012) 768–776.
[54] D. Grassi, C. Lippi, S. Necozione, G. Desideri, C. Ferri, Am. J. Clin. Nutr. 81 (2005) 611–614.
[55] D. Grassi, S. Necozione, C. Lippi, G. Croce, L. Valeri, P. Pasqualetti, G. Desideri, J.B. Blumberg, C. Ferri, Hypertension 46 (2005) 398–405.
[56] D. Grassi, G. Desideri, S. Necozione, C. Lippi, R. Casale, G. Properzi, J.B. Blumberg, C. Ferri, J. Nutr. 138 (2008) 1671–1676.
[57] G. Desideri, C. Kwik-Uribe, D. Grassi, S. Necozione, L. Ghiadoni, D. Mastroiacovo, A. Raffaele, L. Ferri, R. Bocale, M.C. Lechiara, C. Marini, C. Ferri, Hypertension 60 (2012) 794–801.
[58] L. Hooper, C. Kay, A. Abdelhamid, P.A. Kroon, J.S. Cohn, E.B. Rimm, A. Cassidy, Am. J. Clin. Nutr. 95 (2012) 740–751.
[59] K. Davison, A.M. Coates, J.D. Buckley, P.R.C. Howe, Int. J. Obes. 32 (2008) 1289–1296.
[60] D. Esser, J.M. Geleijnse, J.C. Matualatupauw, J.I. Dower, D. Kromhout, P.C.H. Hollman, L.A. Afman, PLoS One 13 (2018) e0194229.
[61] A.M. Ramírez-Rodríguez, M. González-Ortiz, E. Martínez-Abundis, N. Acuña Ortega, J. Med. Food 20 (2017) 882–886.
[62] Y. Jia, S. Kim, J. Kim, B. Kim, C. Wu, J.H. Lee, H.-J. Jun, N. Kim, D. Lee, S.-J. Lee, Mol. Nutr. Food Res. 59 (2015) 344–354.
[63] A. Sundaresan, T. Radhiga, K.V. Pugalendi, J. Physiol. Biochem. 72 (2016) 345–352.
[64] Z. Samadi Noshahr, M.R. Shahraki, H. Ahmadvand, D. Nourabadi, A. Nakhaei, Rep Biochem Mol Biol 3 (2015) 62–67.
[65] T. Anwer, M. Sharma, K.K. Pillai, M. Iqbal, Basic Clin. Pharmacol. Toxicol. 102 (2008) 498–503.
[66] J. Lee, J. Liu, X. Feng, M.A. Salazar Hernández, P. Mucka, D. Ibi, J.W. Choi, U. Ozcan, Nat. Med. 22 (2016) 1023–1032.
[67] U.J. Jung, Y.-Y. Cho, M.-S. Choi, Nutrients 8 (2016).
[68] Shi T., Zhuang R., Zhou H., Wang F., Shao Y., Cai Z., Zhonghua Gan Zang Bing Za Zhi 23 (2015) 124–129.
[69] W.H. Choi, H.J. Son, Y.J. Jang, J. Ahn, C.H. Jung, T.Y. Ha, Mol. Nutr. Food Res. 61 (2017).
[70] V.T.T. Huyen, D.V. Phan, P. Thang, N.K. Hoa, C.G. Ostenson, J. Nutr. Metab. 2013 (2013) 765383.
[71] V.T.T. Huyen, D.V. Phan, P. Thang, P.T. Ky, N.K. Hoa, C.G. Ostenson, Evid. Based. Complement. Alternat. Med. 2012 (2012) 452313.
[72] V.T.T. Huyen, D.V. Phan, P. Thang, N.K. Hoa, C.G. Ostenson, Horm. Metab. Res. 42 (2010) 353–357.
[73] H.J. Choi, J. Yu, H. Choi, J.H. An, S.W. Kim, K.S. Park, H.C. Jang, S.Y. Kim, C.S. Shin, Diabetes Care 34 (2011) e147.
[74] M. Yoshida, P.F. Jacques, J.B. Meigs, E. Saltzman, M.K. Shea, C. Gundberg, B. Dawson-Hughes, G. Dallal, S.L. Booth, Diabetes Care 31 (2008) 2092–2096.
[75] Y. Li, J.P. Chen, L. Duan, S. Li, Diabetes Res. Clin. Pract. 136 (2018) 39–51.
[76] T. Li, S. Chen, T. Feng, J. Dong, Y. Li, H. Li, Food Funct. 7 (2016) 1147–1154.
[77] N. Kamalakkannan, P.S.M. Prince, Basic Clin. Pharmacol. Toxicol. 98 (2006) 97–103.
[78] A.C. Burke, B.G. Sutherland, D.E. Telford, M.R. Morrow, C.G. Sawyez, J.Y. Edwards, M. Drangova, M.W. Huff, J. Lipid Res. 59 (2018) 1714–1728.
[79] Y.-J. Kim, M.-S. Choi, J.T. Woo, M.J. Jeong, S.R. Kim, U.J. Jung, Mol. Nutr. Food Res. 61 (2017).
[80] B. Qin, M.M. Polansky, R.A. Anderson, Horm. Metab. Res. 42 (2010) 187–193.
[81] B. Qin, H.D. Dawson, N.W. Schoene, M.M. Polansky, R.A. Anderson, Nutrition 28 (2012) 1172–1179.
[82] M. Hajimonfarednejad, M. Nimrouzi, M. Heydari, M.M. Zarshenas, M.J. Raee, B.N. Jahromi, Phytother. Res. 32 (2018) 276–283.
[83] M.H. Sharawy, M.S. El-Awady, N. Megahed, N.M. Gameil, Can. J. Physiol. Pharmacol. 94 (2016) 488–497.
[84] W. Liu, S. Zhao, J. Wang, J. Shi, Y. Sun, W. Wang, G. Ning, J. Hong, R. Liu, Mol. Nutr. Food Res. 61 (2017).
[85] H.-J. Zhang, B.-P. Ji, G. Chen, F. Zhou, Y.-C. Luo, H.-Q. Yu, F.-Y. Gao, Z.-P. Zhang, H.-Y. Li, J. Food Sci. 74 (2009) H1–7.
[86] S. Timmers, E. Konings, L. Bilet, R.H. Houtkooper, T. van de Weijer, G.H. Goossens, J. Hoeks, S. van der Krieken, D. Ryu, S. Kersten, E. Moonen-Kornips, M.K.C. Hesselink, I. Kunz, V.B. Schrauwen-Hinderling, E. Blaak, J. Auwerx, P. Schrauwen, Cell Metab. 14 (2011) 612–622.
[87] M. Lagouge, C. Argmann, Z. Gerhart-Hines, H. Meziane, C. Lerin, F. Daussin, N. Messadeq, J. Milne, P. Lambert, P. Elliott, B. Geny, M. Laakso, P. Puigserver, J. Auwerx, Cell 127 (2006) 1109–1122.
[88] P. Brasnyó, G.A. Molnár, M. Mohás, L. Markó, B. Laczy, J. Cseh, E. Mikolás, I.A. Szijártó, A. Mérei, R. Halmai, L.G. Mészáros, B. Sümegi, I. Wittmann, Br. J. Nutr. 106 (2011) 383–389.
[89] M. Malaguarnera, M.P. Gargante, C. Russo, T. Antic, M. Vacante, M. Malaguarnera, T. Avitabile, G. Li Volti, F. Galvano, Am. J. Gastroenterol. 105 (2010) 1338–1345.
[90] A. Molfino, A. Cascino, C. Conte, C. Ramaccini, F. Rossi Fanelli, A. Laviano, JPEN J. Parenter. Enteral Nutr. 34 (2010) 295–299.
[91] B. Capaldo, R. Napoli, P. Di Bonito, G. Albano, L. Saccà, Diabetes Res. Clin. Pract. 14 (1991) 191–195.
[92] P.L. Prieto-Hontoria, P. Pérez-Matute, M. Fernández-Galilea, J. Alfredo Martínez, M.J. Moreno-Aliaga, Eur. J. Nutr. 52 (2013) 779–787.
[93] A. El Midaoui, J. de Champlain, Hypertension 39 (2002) 303–307.
[94] F. Corrado, R. D’Anna, G. Di Vieste, D. Giordano, B. Pintaudi, A. Santamaria, A. Di Benedetto, Diabet. Med. 28 (2011) 972–975.
[95] E. Benelli, S. Del Ghianda, C. Di Cosmo, M. Tonacchera, Int. J. Endocrinol. 2016 (2016) 3204083.
[96] A.D. Genazzani, C. Lanzoni, F. Ricchieri, V.M. Jasonni, Gynecol. Endocrinol. 24 (2008) 139–144.






No Comments Yet
Sign in or Register to Comment