
Key Learning Objectives
- Start to appreciate why redundancy is important in living systems.
- Introduce the three pathways used to make NAD+.
- Discover how L-tryptophan at breakfast influences what happens at night.
- Learn about how L-tryptophan is used to make NAD+.
- Find out which dietary nutrients support making NAD+ from L-tryptophan
- Understand why preventing a build-up of quinolinic acid (QA) is important.
Complex Systems Science and Redundancy
Complexity science is not about one thing in isolation; it’s about interacting networks of things (including our cellular network with mitochondrial networks, and both with the gut microbiome), redundancy, self-regulation, and whole system responses. In this article (the second in our scientific series on NAD+) we’ll be introducing the redundancy in making the NAD+ molecule. Redundancy implies having more than one way to do something. In biological systems it means that, if plan A fails, there’s a backup plan … and probably a backup to the backup. This is exactly what we see with making the NAD+ molecule.
Biological complex adaptive systems often have more than one way to accomplish the same outcome. This allows for robustness, flexibility, and adaptability. These attributes are needed for successfully surviving in the face of real-world challenges. Given the importance of NAD+ in cellular energy production, nutrient sensing, and many other functions, it should be no surprise that there’s more than one way we make it.
NAD+ can be made via the “De Novo Pathway,” starting from the essential amino acid L-tryptophan. It can be made by the “Preiss-Handler pathway,” using nicotinic acid, a form of vitamin B3 usually called niacin, which is known for producing flushing when taken in high amounts. And, it can be produced in the “Salvage Pathway” from niacinamide (nicotinamide), the non-flushing form of vitamin B3. Different tissues use these three pathways to greater or lesser extents to meet changing NAD+ needs. Because, in general, giving more choices allows for better adaptation, it makes sense to support all three pathways.
The type of redundancy that can be used in making NAD+ is called degeneracy. It means that structurally dissimilar components/modules/pathways can perform similar functions (i.e. are effectively interchangeable) under certain conditions, but perform distinct functions in other conditions.
Three Pathways, One Molecule
Because of its overall importance, most organisms have several alternatives for producing the NAD+ molecule. In humans, there are three major NAD+ biosynthesis pathways. These are:

NAD+ can be synthesized de novo through multiple enzymatic steps in the kynurenine pathway (KP), ultimately producing nicotinic acid mononucleotide (NaMN) after the last enzyme step. In biochemistry "de novo" means one biomolecule—NAD+ in this case—is synthesized anew from a different molecule. In this pathway, the niacin molecule is essentially built from scratch starting from the essential amino acid L-tryptophan (Trp). This is the only non-vitamin B3 pathway for producing NAD+. The de novo synthesis pathway is complete with the formation of NaMN.
Niacins (i.e., nicotinic acid- or niacinamide-containing compounds) supplied in the diet and/or supplements can be used to synthesize NAD+, with each form entering NAD+ biosynthesis pathways at different points and often relying on one or more unique enzymes.
Nicotinic acid (NA) and nicotinic acid riboside (NAR) produce NAD+ by the Preiss-Handler pathway. The Preiss–Handler pathway starts from a niacin (either NA or NAR). It proceeds to NAD+ in several enzymatic steps. NaNM is an intermediate in the pathway, so de novo synthesis from Trp shares several steps in this pathway to complete NAD+ synthesis.
A “salvage” pathway converts nicotinamide (NAM; Niacinamide) to NAD+ with nicotinamide mononucleotide (NMN) as an intermediate. It is called a salvage pathway because, no matter what precursor (Trp or any of the niacins) or pathway produces NAD+, after NAD+ is consumed, NAM is generated and is a "salvageable precursor" to re-produce NAD+. So, the salvage pathway is the mechanism used to recycle NAM into NAD+. Nicotinamide riboside (NR) also uses the salvage pathway. Enzymes exist that can convert NR into NAM or NMN. In either instance, NAD+ formed by NR can be used in NAD+-consuming reactions and ends up as NAM. NMN would also be a salvage pathway niacin equivalent.
The NAD+ molecule can be made from the (1) amino acid L-tryptophan, (2) compounds containing a nicotinic acid (i.e., niacin), or (3) compounds containing a nicotinamide (i.e., niacinamide).
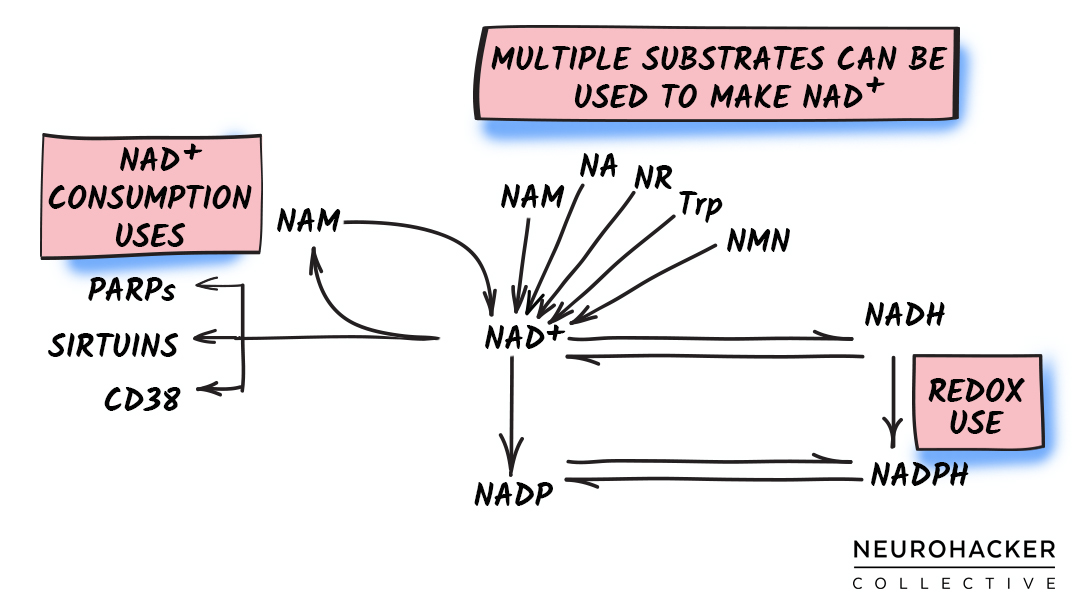 Figure 1. NAD+ Biosynthesis - Major Molecules and Pathway
Figure 1. NAD+ Biosynthesis - Major Molecules and Pathway
Neurohacker Collective’s Complexity Science Approach
Different NAD+ precursor molecules and pathways appear to be more (and less) active in certain tissues. But overall, the three NAD+ biosynthetic routes allow for some degree of functional redundancy.[1] This is important because all humans are not alike, and our capacity for generating NAD+ via each pathway might change in certain tissues because of factors such as genetics, age, and health status.
Aging results in decreased NAD+ and changes to the NAD metabolome—metabolome is a scientific way of saying the metabolites made from and that make the NAD molecule.[2] Alterations in both biosynthesis and consumption appear to stress NAD+ pools in ways that result in insufficient supply to meet the demands of healthy aging.[3] To best support these demands, we believe it’s better to support the functional redundancy created by the three NAD+ biosynthesis pathways. This includes supplying at least one precursor for each pathway and supporting the different pathways with the cofactors needed for enzyme function. It means putting an emphasis on upregulating rate-limiting enzymes. And it might mean, depending on the pathway, nudging flows gently in directions that support higher levels of NAD+ and/or the type of responses seen in younger, healthy persons.
Our complex systems science approach supports all the biosynthesis pathways. We chose this approach because the body is capable of self-regulating NAD+ biosynthesis to meet tissue-specific and subcellular compartment NAD+ demands in ways that science does not completely understand. We believe it can do this more efficiently when it’s being supported comprehensively to create healthy flux through the three different pathways.
We think it’s important to nutritionally support the interconnected NAD+ pathways in ways that create healthy flows of all the molecules in the NAD+ metabolome.
 Figure 2. NAD+ Biosynthesis Support Stack
Figure 2. NAD+ Biosynthesis Support Stack
De Novo Synthesis Pathway Support: Introduction

Dietary L-tryptophan (Trp) (figure 3) is classified as a niacin equivalent in humans because it can be used to generate NAD+ through a de novo synthesis pathway. This pathway forms the pyridine ring common to all niacins, so essentially builds a niacin molecule anew. De novo synthesis starts with Trp and proceeds as this essential amino acid is catabolized through the kynurenine pathway (KP). The KP is a subset of the entire de novo synthesis pathway, with the end-product produced still being an intermediate of NAD+ synthesis.

Figure 3. L-Tryptophan Diagram
In addition to KP, Trp is a precursor molecule for three other pathways. These are (1) hydroxylation (products include serotonin, 5-hydroxy-tryptamine, and melatonin); (2) decarboxylation (products include tryptamine); and (3) transamination (products include indolepyruvic acid). Estimates suggest that ~95% of Trp is degraded through KP, so Trp flux through these other 3 pathways is comparatively minor.[4,5]
L-tryptophan is used to build NAD+ from scratch. This is why the pathway is called ‘de novo,’ which in Latin means anew or from the beginning.
Niacin Equivalent Activity of L-Tryptophan
Trp appears to generally be independent from (so additive to) niacins/niacinamides for NAD+ generation.[6] As an example, in young women, niacinamide (NAM) supplementation did not affect flux through the de novo pathway, with no observed changes in KP intermediates as NAM doses were increased.[7] The additive nature of Trp with NAM suggests that better self-regulatory benefits might be achieved if this pathway is supported along with pathways that use niacin/niacinamide.
It’s been estimated that 60 mg of Trp is the NAD+ equivalent to 1 mg of niacin in men.[8] In Japanese women a conversion ratio of 67 mg Trp to make 1 mg niacin has been reported.[9] Other conversion ratios have also been reported.[10,11] There’s some degree of individual variability, with many factors affecting conversion, including nutrients, hormones (e.g., cortisol, estrogen), pregnancy, drugs, and diseases. Some of these factors enhance, and others suppress the conversion of Trp to NAD+.[12] In niacin-deficient animals, as an example, Trp has about 1/10 (molar ratio) the activity compared to NAM, indicating it is much more efficient in producing NAD+ under these circumstances.[13,14] Supplying enough Trp can correct vitamin B3 deficiency in animals fed a diet devoid of niacins,[12] so a key point is that, despite any individual variance in conversion, Trp is a useful substrate for NAD+ synthesis, and is an especially important substrate under circumstances where tissues need it to be.
While Trp is not thought to contribute a large amount to the overall NAD+ pool under normal circumstances, there are times where its contribution takes on much greater importance.[12] Trp has been suggested to be the primary source of NAD+ during immune system activation.[15] In general, the flux of Trp into the KP increases with age and inflammation.[16–18] During neuroinflammation (at least in a mouse model of Alzheimer’s disease), 95% of the cerebral pool of Trp is catabolized through the KP.[19]
Under some circumstances, aging and inflammation as examples, L-tryptophan takes on much greater importance in making NAD+.
Trp is thought to be a principal NAD+ precursor utilized in the liver, since the liver contains all the enzymes needed for de novo synthesis.[12,20,21] An estimated 90% of Trp degradation by KP occurs in the liver under normal circumstances. In general, flux through the KP pathway (and NAD+ production) in the liver increases dose-dependently with increasing intake of Trp up to about 5 grams.[22–24]
The NAD+ produced from Trp in the liver appears to be used locally, with nicotinamide (NAM), not the NAD+ molecule, released into circulation to be used in peripheral tissues.[12,25] Increasing Trp intake allows more flux through the KP, which increases the amount of NAM released into the vasculature (potentially upregulating the salvage pathway of NAD+ generation).[20,26,27] Conversely, under conditions of low Trp consumption, enzymes that direct Trp to non-NAD+ biosynthetic routes are down-regulated, suggesting a shift of all possible Trp catabolism to NAD+ generation when lower amounts are available in the diet.[27] These findings highlight the importance of supplying modest amounts of Trp for both liver and systemic NAD+ maintenance.
Extrahepatic KP activity accounts for 5-10% of Trp degradation, and becomes more important during circumstances characterized by inflammation and immune activation. Extrahepatic KP does not include all enzymes of the pathway, so some intermediates of the KP are not produced in all tissues.[21,28]
When L-tryptophan (Trp) in the diet is low, non-NAD+ uses of this amino acid are down-regulated, suggesting the body is prioritizing making NAD+ over other possible uses of Trp.
The Kynurenine Pathway (simplified)
The kynurenine pathway (KP) is not a linear pathway. Several intermediates (e.g., kynurenine, 3-hydroxykynurenine [3-HK], 2-amino-3-carboxymuconate-6-semialdehyde [ACMS]) can branch Trp degradation to produce other metabolic intermediates (e.g., anthranilic acid [AA], kynurenic acid [KA], picolinic acid [PA], acetyl CoA) rather than continuing towards NAD+.[29]. The simplified pathway described below will focus on the Trp to NAD+ progression.
While this is “simplified,” it does focus on the biochemistry and uses enzyme names and other scientific terms. This section is written for persons who want to know that level of detail. If that’s not you, it’s okay to skip, but do take a look at anything that is in bold and the quotes in bigger font and italics: those will hit the high notes of this section.
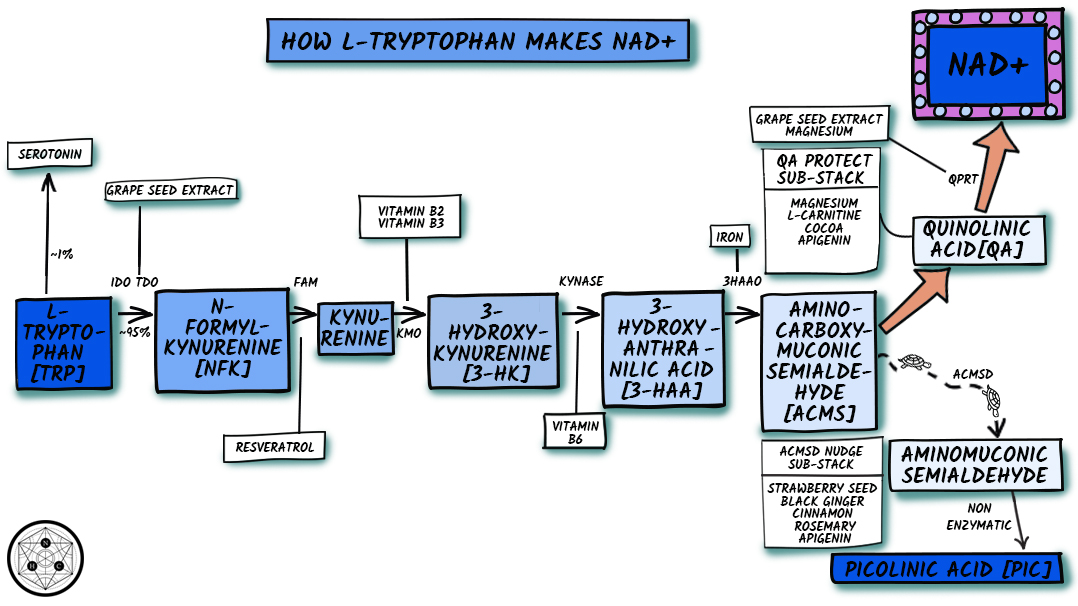
Figure 4. Simplified Kynurenine Pathway
Trp is initially converted to N′-formylkynurenine (NFK) by oxidative degradation using either tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) (both enzymes are heme dependent but are independent of each other). The flux of Trp down the KP is determined primarily by Trp availability, and secondarily by activities of TDO and IDO.
The flux of L-tryptophan (Trp) down the kynurenine pathway (KP) is strongly influenced by how much Trp is available: More available Trp results in greater flow towards NAD+ production.
TDO is expressed mainly in the liver, but also exists and has activity in the brain. It’s responsible for most KP metabolism of Trp and is upregulated by Trp availability and cortisol (as well as other glucocorticoids).[12,17,30] Because of cortisol's robust circadian rhythm, dietary Trp would be expected to be largely shunted into the KP in the morning. This makes biological sense, since, as an example, we don’t need a big increase in Trp being made into melatonin until nighttime.
IDO is widely expressed in all tissues (including blood and immune cells). IDO is thought to have negligible activity under normal conditions, but is upregulated during immune system activation and inflammation. Its activity also tends to increase with aging.
IDO is regulated by both pro- and anti-inflammatory molecules.[31–34] The principle inducer is interferon-gamma (IFN-γ) in the periphery and interleukin-6 (IL-6) in the central nervous system.[35–37] Other proinflammatory molecules including IFN-α, interleukins (IL-1β, IL-2) and tumor necrosis factor α (TNFα) can induce IDO, but do so with less activity.[38] In general, anti-inflammatory cytokines inhibit IDO induction.
Because of this counter-regulation, IDO status is assumed to be determined by the overall balance between pro- and anti-inflammatory cytokines. During circumstances characterized by increased systemic inflammation or brain neuroinflammation, IDO dramatically increases Trp flux through the KP.[30,39–43] It’s been suggested that the higher activity of IDO might be a self-regulatory adaptation intended to increase overall NAD+ synthesis, so more of this molecule would be available for its consuming enzymes (e.g., PARP, CD38, Sirtuins) to support healthier immune and inflammatory responses.[15]
During inflammation or immune system activation, KP, specifically an enzyme abbreviated IDO, is upregulated. This occurs because tissues need more NAD+ during these circumstances, and use L-tryptophan to make it.
Whether produced using TDO or IDO, NFK is converted to kynurenine (Kyn) by NFK formamidase (FAM), which does not have any known cofactors and is not rate-limiting. Kyn is the first stable metabolite formed in the KP. Very high doses of resveratrol (5 g/d) have increased Kyn in humans (and decreased Trp presumably because more of it is entering the KP).[44] This study did not measure enzyme activity, but since resveratrol down-regulates IFN-γ and pro-inflammatory activation of KP via IDO in vitro,[45] it’s possible that resveratrol upregulated TDO and/or FAM.
The next two steps are dependent on specific B vitamins (which have been included in the stack). Hydroxylation by kynurenine hydroxylase (K monooxygenase [KMO]) forms 3-HK. KMO uses the reduced NADPH coenzyme form of niacin. NADPH is a flavin adenine dinucleotide–dependent enzyme. Because of the flavin dependence, activity of KMO is diminished several-fold during vitamin B2 (riboflavin) deficiency.[46,47]
3-HT is converted to 3-hydroxyanthranilic acid (3-HAA) by kynureninase B (KYNase)—a pyridoxal 5′-phosphate (P5P) (vitamin B6) dependent enzyme. KYNase activity is impaired significantly by vitamin B6 deficiency.[46,48–53] Drugs, especially estrogen-containing therapies (e.g., oral contraceptives, hormone replacement therapy) can stress vitamin B6 status and also decrease KYNase activity.[54–57]
While deficiency of either vitamin B2 or B6 will inhibit flux through the KP, large intakes are not required to support these enzymes. Supplying an amount in the range of daily values (DV) has ensured that these enzymes are not rate-limiting, while significantly higher vitamin B2 and B6 have not further increased formation of KP intermediates formed after these enzymes.[57–60] Because of this, we think it’s prudent to supply approximately the DV amount of these vitamins to ensure adequacy, but high dosing does not appear warranted for KP function.
Vitamin B2 and B6 are used as cofactors in the KP. Ensuring daily values of both are being supplied is important to keep good flow through this pathway.
The next step uses 3-hydroxyanthranilic acid 3,4-dioxygenase (3-HAAO) to convert 3-HAA to ACMS. 3-HAAO is a non-heme iron-dependent (Fe2+) dioxygenase enzyme; it is considered the most active among KP enzymes, rapidly converting 3-HAA into ACMS. So under most circumstances the activity of this enzyme is not an issue. The exception might be during iron deficiency. Iron deficiency has reduced Trp utilization for NAD+ synthesis in animals (presumably because of this enzyme),[61] and relative availability of endogenous iron and proteins that regulate its bioavailability appear to impact activity of 3-HAAO.[62] Our intention is that this stack will be taken long-term. We don’t think it’s prudent to supplement iron for long periods of time unless iron status is being monitored. Because of this, we did not include iron in the stack. In persons who are iron-deficient, correcting iron status might be an important part of optimizing de novo synthesis of NAD+.
ACMS is an unstable intermediate (i.e., it spontaneously gets converted into other molecules) and does not require enzyme activity to convert to quinolinic acid (QA).[10] But, only about 6% of ACMS is spontaneously condensed and rearranged into QA.[63] The majority of ACMS is believed to be shunted away from NAD+ production (towards picolinic acid and acetyl-CoA synthesis) by α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD).[64,65] ACMSD is zinc-dependent.[66] Change in zinc availability has been proposed to influence its activity.[67] ACMSD inhibition (i.e., slowing of activity) nudges more ACMS towards QA and hence NAD+ biosynthesis,[68] while overactivity of ACMSD reduces QA.[69] Increased activity of PPARα down-regulates ACMSD.[70] While not major players in this stack, several of our ingredient choices in the overall formula have upregulated PPARα in pre-clinical studies, so might theoretically nudge pathway flux more towards QA production (and hence in the NAD+ direction). These ingredients include: (1) strawberry seed (standardized for trans-tiliroside),[71] (2) Kaempferia parviflora (black ginger),[72] (3) rosemary extract (standardized for ursolic acid),[73,74] (4) cinnamon,[75] and (5) apigenin.[76,77]
One of our core formulation principles is to have more than one ingredient that might do something and to include ingredients that do more than one thing.
QA production ends the KP portion of de novo synthesis. The next enzymatic step converts QA to a niacin-containing molecule. This step uses quinolinate phosphoribosyl transferase (QPRT) to create Nicotinic Acid Mononucleotide (NaMN)—magnesium and 5-phosphoribosyl-1-pyrophosphate (PRPP) are cosubstrates.[27] NaMN converges with the Preiss-Handler pathway (discussed in the next article in this series): It is a common intermediate in both pathways and both pathways share the same enzymes steps to make NAD+ from this point.
Trp that makes it to QA in the liver is thought to be converted to NAD+ via subsequent enzyme reactions of QPRT and the Preiss-Handler pathway, since all needed enzymes are highly expressed in liver tissue.[78,79] The brain is also a big producer of NAD+ from QA.
QA might be best thought of as a “Goldilocks” molecule—we need some of it, but it is an excitatory molecule, so too much is neurotoxic to nerve cells in the brain. QPRT is expressed in the brain; it plays a major role in protection against the neurotoxic effects of quinolinic acid.[78,80–82] QPRT activity can increases in response to increased levels of QA,[83] suggesting a protective role (i.e., the QPRT enzyme is being upregulated in response to QA to make sure this molecule isn’t allowed to build-up).
The last step in the KP on the journey to NAD+ takes an intermediate called quinolinic acid (QA) and converts it into a niacin-containing compound using an enzyme abbreviated QPRT. This is an important step to support, because we don’t want QA to build-up (it can damage neurons if too much accumulates).
The activity of QPRT can act as a rate-limiting factor for NAD+ generation from Trp.[63] In other words, the enzymes in the KP that make QA from Trp do their jobs, but QPRT can’t keep pace. The result is that QA accumulates. This commonly occurs in tissues, especially the brain and nervous system, during circumstances characterized by inflammation (like the chronic inflammation of aging called “inflammaging”) and immune activation. As an example, although QPRT activity is known to upregulate in response to increases in the levels of QA, its capacity is insufficient to keep up with the QA being produced in circumstances characterized by neuroinflammation, resulting in a build-up of QA.[19,83–96] A similar finding occurs in plasma, where chronic inflammatory circumstances are associated with increased QA.[15,97–99] The highest levels of QA are found in spleen, lymph nodes, thymus, and many specific immune cell types and are increased following stimulation by immune activators.[100] Higher QA levels have also been reported in cardiovascular disease.[101] The key point is that QPRT activity is essential for preventing a build-up of QA.
Activity level of the de novo synthesis pathway is impacted with age,[18,102–106] but in tissue-specific ways.[17] Activity of TDO (the non-inflammation first enzyme in the pathway) decreases significantly with age progression in the brain, liver and kidney. Activity of the inflammation-inducible IDO increases with age in the brain, but decreases in the liver and kidney. Activity of QPRT declines in the brain and liver, but increases in kidneys.[105] Because of the critical importance of QPRT in generating the end-product of the KP (NaMN), the overall pathway performance is downregulated during aging in two tissues that heavily rely on it (e.g., brain, liver). Not surprisingly, in humans, QA levels are positively correlated with age (i.e., the amount of QA increases as we get older).[18,106]
Some enzymes involved in making NAD+ from L-tryptophan decrease with age in tissues like the brain and liver. This affects our ability to maintain healthy pools of NAD+.
Reduced ability to convert QA to NaMN is an issue for several reasons. In certain tissues and cells (e.g., neurons, astrocytes), and under some circumstances (e.g., neuroinflammation, immune system activation), NAD+ production from QA (and hence Trp) appears to be at least, if not more important than that from niacinamide (NAM) (and the salvage pathway) or nicotinic acid (NA) (and the Preiss–Handler pathway).[39,93,107] It’s thought that KP activation is upregulated during neuroinflammation in an attempt to maintain NAD+ levels through de novo synthesis from Trp. If QPRT is unable to effectively convert QA to NaMN, then NAD+ levels will be inadequate to meet cellular needs and tissue demands: the goal of the upregulation will not be met.
In addition to inefficient generation of NAD+, QA itself can be health-damaging. QA is an NMDA agonist and exerts excitotoxic effects on neurons.[108–110] So elevated QA is not only associated with many neuroinflammatory conditions, it is believed to be contributing to worsening them.
Inhibiting the KP—specifically IDO and QPRT—during neuroinflammatory circumstances might solve one problem (the build-up of QA), but cause others. Competitive inhibition of IDO and QPRT activities causes a dose-dependent decrease in intracellular NAD+ levels and sirtuin deacetylase-1 (SIRT1) activity in astrocytes and neurons (neurons are more affected than astrocytes): this results in impaired cell viability.[93]
Because of these reasons, supporting upregulation of QPRT activity and protecting against QA’s health-damaging effects are important. Grape seed proanthocyanidin dose-dependently and robustly increased liver NAD+ levels and sirtuin activity in healthy rats. This was in part a result of increased KP activity. Supplementation increased TDO and also dose-dependently upregulated QPRT, with effects appearing at the lowest dose (comparable to about 56 mg for a 70 kg human adult).[111]
Magnesium acts as core support because it is both a cosubstrate for QPRT and protects neuronal cells against QA toxicity.[112–115] In experiments, higher brain QA leads to increased Mg uptake into the brain,[116] suggesting that if Mg is available in circulation under circumstances characterized by higher brain QA levels, it will be put to good use.
Several other ingredients in the stack have neuroprotective benefits against QA. One of these is L-carnitine.[117,118] Certain polyphenol molecules, including catechin (found in cocoa extract) and apigenin have neuroprotective effects against QA and prevent QA-mediated NAD+ depletion in cultured human neurons.[119]
The last part of the stack has to do with experimental findings indicating that insufficient mineral intake decreased the conversion rate of Trp to NAD+ in rats.[12] As previously mentioned several enzymes in the KP are dependent on minerals (e.g., iron, magnesium, and zinc). The stack contains 70 trace minerals (found in the peat extract component of elevATP®), including these three minerals.
We believe a comprehensive NAD+ stack should offer full pathway support of the KP. This means (1) supplying a modest amount of Trp, (2) including vitamin coenzymes and other nutritional cosubstrates needed to support enzyme function, and (3) and supporting activity of the enzyme step that acts as a functional rate-limited enzyme (QPRT).
References
[1] A. Rongvaux, F. Andris, F. Van Gool, O. Leo, Bioessays 25 (2003) 683–690.
[2] J. Clement, M. Wong, A. Poljak, P. Sachdev, N. Braidy, Rejuvenation Res. (2018).
[3] S. Johnson, S.-I. Imai, F1000Res. 7 (2018) 132.
[4] D.A. Bender, Mol. Aspects Med. 6 (1983) 101–197.
[5] A.A. Badawy, Adv. Exp. Med. Biol. 467 (1999) 265–274.
[6] M. Shin, S. Nakakita, C. Hashimoto, K. Sano, C. Umezawa, Int. J. Vitam. Nutr. Res. 68 (1998) 104–108.
[7] T. Fukuwatari, K. Shibata, Int. J. Vitam. Nutr. Res. 77 (2007) 255–262.
[8] M.K. Horwitt, C.C. Harvey, W.S. Rothwell, J.L. Cutler, D. Haffron, J. Nutr. 60 (1956) 1–43.
[9] T. Fukuwatari, M. Ohta, N. Kimura, R. Sasaki, K. Shibata, J. Nutr. Sci. Vitaminol. 50 (2004) 385–391.
[10] G.A. Goldsmith, Am. J. Clin. Nutr. 6 (1958) 479–486.
[11] I. Nakagawa, T. Takahashi, T. Suzuki, Y. Masana, J. Nutr. 99 (1969) 325–330.
[12] T. Fukuwatari, K. Shibata, Int. J. Tryptophan Res. 6 (2013) 3–8.
[13] W.A. Krehl, D. Bonner, C. Yanofsky, J. Nutr. 41 (1950) 159–172.
[14] K. Shibata, M. Swabe, T. Fukuwatari, E. Sugimoto, Biosci. Biotechnol. Biochem. 64 (2000) 206–209.
[15] J.R. Moffett, M.A. Namboodiri, Immunol. Cell Biol. 81 (2003) 247–265.
[16] B. Frick, K. Schroecksnadel, G. Neurauter, F. Leblhuber, D. Fuchs, Clin. Biochem. 37 (2004) 684–687.
[17] Y. Chen, G.J. Guillemin, Int. J. 2 (2009) IJTR.S2097.
[18] J. de Bie, J. Guest, G.J. Guillemin, R. Grant, J. Neurochem. 136 (2016) 995–1003.
[19] W. Wu, J.A. Nicolazzo, L. Wen, R. Chung, R. Stankovic, S.S. Bao, C.K. Lim, B.J. Brew, K.M. Cullen, G.J. Guillemin, PLoS One 8 (2013) e59749.
[20] D.A. Bender, B.I. Magboul, D. Wynick, Br. J. Nutr. 48 (1982) 119–127.
[21] T. Fukuwatari, Y. Morikawa, E. Sugimoto, K. Shibata, Biosci. Biotechnol. Biochem. 66 (2002) 1196–1204.
[22] K. Shibata, H. Matsuo, J. Nutr. 120 (1990) 1191–1197.
[23] C. Hiratsuka, T. Fukuwatari, M. Sano, K. Saito, S. Sasaki, K. Shibata, J. Nutr. 143 (2013) 859–866.
[24] A.A.-B. Badawy, D.M. Dougherty, Int. J. Tryptophan Res. 9 (2016) 31–49.
[25] L. Liu, X. Su, W.J. Quinn 3rd, S. Hui, K. Krukenberg, D.W. Frederick, P. Redpath, L. Zhan, K. Chellappa, E. White, M. Migaud, T.J. Mitchison, J.A. Baur, J.D. Rabinowitz, Cell Metab. 27 (2018) 1067–1080.e5.
[26] G.W. Beadle, H.K. Mitchell, J.F. Nyc, Proc. Natl. Acad. Sci. U. S. A. 33 (1947) 155–158.
[27] D.A. Bender, R. Olufunwa, Br. J. Nutr. 59 (1988) 279–287.
[28] K. Shibata, K. Motooka, K. Kurata, J. Nutr. Sci. Vitaminol. 28 (1982) 11–19.
[29] A.A.-B. Badawy, Biosci. Rep. 35 (2015).
[30] G. Magni, A. Amici, M. Emanuelli, N. Raffaelli, S. Ruggieri, in: D.L. Purich (Ed.), Advances in Enzymology and Related Areas of Molecular Biology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1999, pp. 135–182.
[31] Y. Nishizuka, O. Hayaishi, J. Biol. Chem. 238 (1963) 3369–3377.
[32] A. Chiarugi, R. Carpenedo, M.T. Molina, L. Mattoli, R. Pellicciari, F. Moroni, J. Neurochem. 65 (2002) 1176–1183.
[33] A.A.-B. Badawy, Alcohol Alcohol 49 (2014) 238–250.
[34] A.A.-B. Badawy, Neuropharmacology 112 (2017) 248–263.
[35] E.R. Pfefferkorn, S. Rebhun, M. Eckel, J. Interferon Res. 6 (1986) 267–279.
[36] E. Werner, G. Werner-Felmayer, CDM 8 (2007) 201–203.
[37] T. Samikkannu, Z.M. Saiyed, K.V.K. Rao, D.K. Babu, J.W. Rodriguez, M.N. Papuashvili, M.P.N. Nair, AIDS Res. Hum. Retroviruses 25 (2009) 329–335.
[38] A. Boasso, G. Shearer, CDM 8 (2007) 217–223.
[39] Y. Ozaki, M.P. Edelstein, D.S. Duch, Biochem. Biophys. Res. Commun. 144 (1987) 1147–1153.
[40] R.S. Grant, H. Naif, S.J. Thuruthyil, N. Nasr, T. Littlejohn, O. Takikawa, V. Kapoor, J. Virol. 74 (2000) 4110–4115.
[41] A. Boasso, J.-P. Herbeuval, A.W. Hardy, S.A. Anderson, M.J. Dolan, D. Fuchs, G.M. Shearer, Blood 109 (2007) 3351–3359.
[42] A.A.-B. Badawy, Int. J. Tryptophan Res. 10 (2017) 1178646917691938.
[43] M. Kanai, H. Funakoshi, H. Takahashi, T. Hayakawa, S. Mizuno, K. Matsumoto, T. Nakamura, Mol. Brain 2 (2009) 8.
[44] G.A. Gualdoni, D. Fuchs, G.J. Zlabinger, J.M. Gostner, Pharmacol. Rep. 68 (2016) 1065–1068.
[45] B. Wirleitner, K. Schroecksnadel, C. Winkler, H. Schennach, D. Fuchs, Immunol. Lett. 100 (2005) 159–163.
[46] Z.H.M. Verjee, Int. J. Biochem. 2 (1971) 711–718.
[47] D.A. Bender, in: G.A. Filippini, C.V.L. Costa, A. Bertazzo (Eds.), Recent Advances in Tryptophan Research, Springer US, Boston, MA, 1996, pp. 565–569.
[48] W.E. Knox, Biochem. J 53 (1953) 379–385.
[49] N. Ogasawara, Y. Hagino, Y. Kotake, J. Biochem. 52 (1962) 162–166.
[50] F. Takeuchi, Y. Shibata, Biochem. J 220 (1984) 693–699.
[51] D.A. Bender, E.N.M. Njagi, P.S. Danielian, Br. J. Nutr. 63 (1990) 27–36.
[52] K. Shibata, M. Mushiage, T. Kondo, T. Hayakawa, H. Tsuge, Biosci. Biotechnol. Biochem. 59 (1995) 2060–2063.
[53] J.L. van de Kamp, A. Smolen, Pharmacol. Biochem. Behav. 51 (1995) 753–758.
[54] D.A. Bender, Biochem. Pharmacol. 29 (1980) 707–712.
[55] D.A. Bender, A.E. Laing, J.A. Vale, L. Papadaki, M. Pugh, Biochem. Pharmacol. 32 (1983) 843–848.
[56] D.A. Bender, L. Totoe, Br. J. Nutr. 51 (1984) 219–224.
[57] L. Rios-Avila, B. Coats, Y.-Y. Chi, Ø. Midttun, P.M. Ueland, P.W. Stacpoole, J.F. Gregory 3rd, J. Nutr. 145 (2015) 87–95.
[58] J.I. Patterson, R.R. Brown, H. Linkswiler, A.E. Harper, Am. J. Clin. Nutr. 33 (1980) 2157–2167.
[59] L.V. Hankes, M. Schmaeler, C.R. Jansen, R.R. Brown, in: G. Huether, W. Kochen, T.J. Simat, H. Steinhart (Eds.), Tryptophan, Serotonin, and Melatonin: Basic Aspects and Applications, Springer US, Boston, MA, 1999, pp. 283–287.
[60] K. Shibata, J. Hirose, T. Fukuwatari, Int. J. Tryptophan Res. 8 (2015) 31–39.
[61] E.K. Stachowski, R. Schwarcz, J. Neural Transm. 119 (2012) 123–131.
[62] G.W. Oduho, Y. Han, D.H. Baker, J. Nutr. 124 (1994) 444–450.
[63] K. Shibata, J. Nutr. Sci. Vitaminol. 64 (2018) 90–98.
[64] S.-I. Fukuoka, K. Ishiguro, K. Yanagihara, A. Tanabe, Y. Egashira, H. Sanada, K. Shibata, J. Biol. Chem. 277 (2002) 35162–35167.
[65] T. Li, H. Iwaki, R. Fu, Y. Hasegawa, H. Zhang, A. Liu, Biochemistry 45 (2006) 6628–6634.
[66] D. Martynowski, Y. Eyobo, T. Li, K. Yang, A. Liu, H. Zhang, Biochemistry 45 (2006) 10412–10421.
[67] K. Shibata, T. Fukuwatari, J. Nutr. Sci. Vitaminol. 60 (2014) 334–339.
[68] R. Pellicciari, P. Liscio, N. Giacchè, F. De Franco, A. Carotti, J. Robertson, L. Cialabrini, E. Katsyuba, N. Raffaelli, J. Auwerx, J. Med. Chem. 61 (2018) 745–759.
[69] L. Brundin, C.M. Sellgren, C.K. Lim, J. Grit, E. Pålsson, M. Landén, M. Samuelsson, K. Lundgren, P. Brundin, D. Fuchs, T.T. Postolache, L. Traskman-Bendz, G.J. Guillemin, S. Erhardt, Transl. Psychiatry 6 (2016) e865.
[70] H. Matsuda, R.-T. Gomi, S. Hirai, Y. Egashira, Biosci. Biotechnol. Biochem. 77 (2013) 1416–1419.
[71] T. Goto, A. Teraminami, J.-Y. Lee, K. Ohyama, K. Funakoshi, Y.-I. Kim, S. Hirai, T. Uemura, R. Yu, N. Takahashi, T. Kawada, J. Nutr. Biochem. 23 (2012) 768–776.
[72] H. Kobayashi, E. Horiguchi-Babamoto, M. Suzuki, H. Makihara, H. Tomozawa, M. Tsubata, T. Shimada, K. Sugiyama, M. Aburada, J. Nat. Med. 70 (2016) 54–61.
[73] Y. Jia, S. Kim, J. Kim, B. Kim, C. Wu, J.H. Lee, H.-J. Jun, N. Kim, D. Lee, S.-J. Lee, Mol. Nutr. Food Res. 59 (2015) 344–354.
[74] Y. Zhang, C. Song, H. Li, J. Hou, D. Li, Mol. Med. Rep. 13 (2016) 5309–5316.
[75] X. Sheng, Y. Zhang, Z. Gong, C. Huang, Y.Q. Zang, PPAR Res. 2008 (2008) 581348.
[76] Shi T., Zhuang R., Zhou H., Wang F., Shao Y., Cai Z., Zhonghua Gan Zang Bing Za Zhi 23 (2015) 124–129.
[77] F. Wang, J.-C. Liu, R.-J. Zhou, X. Zhao, M. Liu, H. Ye, M.-L. Xie, Chem. Biol. Interact. 275 (2017) 171–177.
[78] E. Okuno, R. Schwarcz, Biochimica et Biophysica Acta (BBA) - General Subjects 841 (1985) 112–119.
[79] F.S. Fernando, L. Conforti, S. Tosi, A.D. Smith, M.P. Coleman, Gene 284 (2002) 23–29.
[80] M. Wolfensberger, U. Amsler, M. Cuénod, A.C. Foster, W.O. Whetsell Jr, R. Schwarcz, Neurosci. Lett. 41 (1983) 247–252.
[81] S. Feldblum, A. Rougier, H. Loiseau, P. Loiseau, F. Cohadon, P.L. Morselli, K.G. Lloyd, Epilepsia 29 (1988) 523–529.
[82] C. Ko¨hler, L.G. Eriksson, E. Okuno, R. Schwarcz, Neuroscience 27 (1988) 49–76.
[83] A.C. Foster, W.O. Whetsell, E.D. Bird, R. Schwarcz, Brain Res. 336 (1985) 207–214.
[84] M.P. Heyes, Adv. Exp. Med. Biol. 398 (1996) 125–129.
[85] P. Guidetti, P. Hemachandra Reddy, D.A. Tagle, R. Schwarcz, Neurosci. Lett. 283 (2000) 233–235.
[86] T.W. Stone, Prog. Neurobiol. 64 (2001) 185–218.
[87] G.J. Guillemin, B.J. Brew, Redox Rep. 7 (2002) 199–206.
[88] G.J. Guillemin, B.J. Brew, C.E. Noonan, O. Takikawa, K.M. Cullen, Neuropathol. Appl. Neurobiol. 31 (2005) 395–404.
[89] P. Guidetti, R. Schwarcz, in: G. Allegri, C.V.L. Costa, E. Ragazzi, H. Steinhart, L. Varesio (Eds.), Developments in Tryptophan and Serotonin Metabolism, Springer US, Boston, MA, 2003, pp. 137–145.
[90] G.J. Guillemin, G. Smythe, O. Takikawa, B.J. Brew, Glia 49 (2005) 15–23.
[91] G.J. Guillemin, V. Meininger, B.J. Brew, Neurodegener. Dis. 2 (2005) 166–176.
[92] I. Wonodi, R. Schwarcz, Schizophr. Bull. 36 (2010) 211–218.
[93] N. Braidy, G.J. Guillemin, R. Grant, Int. J. Tryptophan Res. 4 (2011) 29–37.
[94] B.M. Campbell, E. Charych, A.W. Lee, T. Möller, Front. Neurosci. 8 (2014) 12.
[95] L.M. Giil, Ø. Midttun, H. Refsum, A. Ulvik, R. Advani, A.D. Smith, P.M. Ueland, J. Alzheimers. Dis. 60 (2017) 495–504.
[96] K.-H. Chang, M.-L. Cheng, H.-Y. Tang, C.-Y. Huang, Y.-R. Wu, C.-M. Chen, Mol. Neurobiol. 55 (2018) 6319–6328.
[97] J.R. Moffett, M.G. Espey, S.J. Gaudet, M.A.A. Namboodiri, Brain Res. 623 (1993) 337–340.
[98] M.F. Murray, Lancet Infect. Dis. 3 (2003) 644–652.
[99] P. Bipath, P.F. Levay, M. Viljoen, BMC Infect. Dis. 15 (2015) 346.
[100] M. Valle, R.W. Price, A. Nilsson, M. Heyes, D. Verotta, Brain 127 (2004) 1047–1060.
[101] H. Mangge, I. Stelzer, E.Z. Reininghaus, D. Weghuber, T.T. Postolache, D. Fuchs, Curr. Med. Chem. 21 (2014) 1931–1937.
[102] R. Yoshida, T. Nukiwa, Y. Watanabe, M. Fujiwara, F. Hirata, O. Hayaishi, Arch. Biochem. Biophys. 203 (1980) 343–351.
[103] R.J.W. Truscott, A.J. Elderfield, Clin. Sci. 89 (1995) 591–599.
[104] S. Comai, C.V.L. Costa, E. Ragazzi, A. Bertazzo, G. Allegri, Clin. Chim. Acta 360 (2005) 67–80.
[105] N. Braidy, G.J. Guillemin, H. Mansour, T. Chan-Ling, R. Grant, FEBS J. 278 (2011) 4425–4434.
[106] L. Capuron, S. Schroecksnadel, C. Féart, A. Aubert, D. Higueret, P. Barberger-Gateau, S. Layé, D. Fuchs, Biol. Psychiatry 70 (2011) 175–182.
[107] N. Braidy, R. Grant, S. Adams, B.J. Brew, G.J. Guillemin, Neurotox. Res. 16 (2009) 77–86.
[108] T.W. Stone, J.H. Connick, J.I. Addae, D.A.S. Smith, P.A. Brooks, in: P.J. Roberts, J. Storm-Mathisen, H.F. Bradford (Eds.), Excitatory Amino Acids, Palgrave Macmillan UK, London, 1986, pp. 367–380.
[109] R.S. Grant, V. Kapoor, J. Neurochem. 70 (2002) 1759–1763.
[110] A. Rahman, K. Ting, K.M. Cullen, N. Braidy, B.J. Brew, G.J. Guillemin, PLoS One 4 (2009) e6344.
[111] G. Aragonès, M. Suárez, A. Ardid-Ruiz, M. Vinaixa, M.A. Rodríguez, X. Correig, L. Arola, C. Bladé, Sci. Rep. 6 (2016) 24977.
[112] S.R. El-Defrawy, R.J. Boegman, K. Jhamandas, R.J. Beninger, Can. J. Physiol. Pharmacol. 64 (1986) 369–375.
[113] G. Wolf, G. Keilhoff, S. Fischer, P. Hass, Neurosci. Lett. 117 (1990) 207–211.
[114] A. Schurr, C.A. West, B.M. Rigor, Brain Res. 568 (1991) 199–204.
[115] H. Xiao, C. Yang, Y. He, N. Zheng, J. Huazhong Univ. Sci. Technolog. Med. Sci. 30 (2010) 397–402.
[116] F. Rothe, G. Wolf, S. Fischer, P. Hass, G. Keilhoff, K. Abicht, Neuroreport 4 (n.d.) 205–207.
[117] D. Silva-Adaya, V. Pérez-De La Cruz, M.N. Herrera-Mundo, K. Mendoza-Macedo, J. Villeda-Hernández, Z. Binienda, S.F. Ali, A. Santamaría, J. Neurochem. 105 (2008) 677–689.
[118] D. Elinos-Calderón, Y. Robledo-Arratia, V. Pérez-De La Cruz, J. Pedraza-Chaverrí, S.F. Ali, A. Santamaría, Exp. Brain Res. 197 (2009) 287–296.
[119] N. Braidy, R. Grant, S. Adams, G.J. Guillemin, FEBS J. 277 (2010) 368–382.






No Comments Yet
Sign in or Register to Comment